Increasing the resolution of measurement has always revolutionized fields; for example, the incredible scientific impact of the invention of microscope and telescope. Single-cell analysis is a good example of a similar revolution unfolding in biology. The human body is made up of nearly 40 trillion cells. Historically, these cells have been examined in bulk, sometimes millions of cells at a time, which cannot capture the differences across cells. Single-cell analysis looks at the individuality of cells. It is beginning to unravel the mystery of cell differentiation by identifying novel cell types, revealing mechanisms that make these cells different from each other, and demonstrating how cells respond to certain diseases or drugs. This relatively new field is already showing immense potential for biological discoveries with applications ranging from cancer to Covid-19 related research.
The amount of single-cell data is increasing at a rapid pace, thanks to the advancement of data measurement technologies. The size of individual datasets is increasing at a similar rate. Analysis of this data typically involves running a data science pipeline. Because the steps of the pipeline are often repeated with changes in parameters, it helps to have an interactive pipeline that can run in near real-time.
ScRNA-seq Analysis of 1.3 Million Mouse Cells in Just Seven and Half Minutes on a Single Intel® Xeon® Processor
There are many kinds of single-cell analyses studying various aspects of cell-differentiation. Single-cell RNA-seq (scRNA-seq) analysis studies the differences in gene expression profiles across cells. It relies on single-cell RNA sequencing, which is an advanced technique that enables measurement of the gene expression of individual cells.
A typical workflow to do scRNA-seq analysis begins with a matrix that consists of the expression levels of the genes in each cell (Figure 1). In the data preprocessing steps, noise is filtered out and the data is normalized to obtain the activity of every human gene in each individual cell of the dataset. During this step, machine learning is often used to correct artifacts from data collection. Subsequently, dimensionality reduction is performed followed by clustering to group cells with similar genetic activity, and visualization of the clusters. With over 800,000 downloads, Scanpy is one of the most widely used toolkits for this analysis.

Figure 1. Pipeline showing the steps in analysis of single-cell RNA sequencing data, starting from gene activity matrix to visualization of different cell clusters.
For a dataset consisting of 1.3 million mouse brain cells, the pipeline depicted in Figure 1 would normally take nearly 5 hours on a single CPU instance (n1-highmem-64) on Google Cloud Platform* (GCP*) using the off-the-shelf (baseline) Scanpy implementation. For the same pipeline, NVIDIA has reported an end-to-end runtime of 686 seconds on a single NVIDIA A100 GPU using NVIDIA RAPIDS*.
At Intel Labs, we collaborated with the Intel® oneAPI Data Analytics Library (oneDAL) team and Katana Graph, to accelerate the pipeline using better parallel algorithms and tuning the performance to the underlying architecture. While this is still a work-in-progress, Table 1 and Figure 2 report our current performance and cloud usage costs. These results were recently presented at Intel Investor Day 2022. The whole pipeline can now be finished on the same single CPU instance (n1-highmem-64) on GCP in just 626 seconds. This performance only gets better with the newer n2 instance types running 3rd Generation Intel® Xeon® Scalable Processors (Ice Lake). We also reduced the memory requirement of the pipeline so that we can use the low-memory n2-highcpu-64 instances instead of high-memory n2-highmem-64 instances. On a single instance of n2-highcpu-64 on GCP, the whole pipeline finishes in just 459 seconds (7.65 minutes). This is nearly 40x faster than the 5-hour baseline that we started with. This is also nearly 1.5x faster than NVIDIA* A100 performance.
The speed-up and reduction in memory requirement has resulted in significant reduction in cloud computing costs (Table 1). The n2-highcpu-64 instance on GCP costs only $0.29. This is nearly 66x cheaper than n1-highmem-64 running baseline Scanpy and 2.4x cheaper than NVIDIA A100 GPU.

Table 11. Execution time and cloud costs for scRNA-seq analysis of 1.3 million mouse brain cells on various GCP instances. The first two columns report published execution time and cloud costs of baseline Scanpy on a single CPU instance (n1-highmem-64) and GPU-accelerated Scanpy on a single GPU instance (a2-highgpu-1g). The last three columns report measured2 execution time and cloud costs of CPU-accelerated Scanpy on single instances of two generations of CPU instance types (n1-highmem-64, n1-highmem-64 and n2-highcpu-64).
Intel & Google Cloud Provide Turnkey, Optimized Solution for HPC Workloads
July 6, 2022 | Intel® oneAPI Base Toolkit, Intel® oneAPI HPC Toolkit
Intel and Google are working together to drive high-performance computing forward on Google Cloud with the release of the Cloud HPC Toolkit. This new resource provides access to tools from the Intel® oneAPI Base and HPC Toolkits—including Intel® MPI Library and Intel® oneAPI Math Kernel Library—to optimize performance through Intel® Select Solutions for Simulations & Modeling. These new tools improve compile times and speed of results and offer multi-vendor acceleration in SYCL.
Why It’s Important
In a nutshell, the new Toolkit simplifies adoption of robust high-performance cloud computing by removing the challenges inherent in groking and overcoming unfamiliar development concepts and tools. (These can result in slow deployment for demanding workloads, software incompatibilities, and subpar performance.)
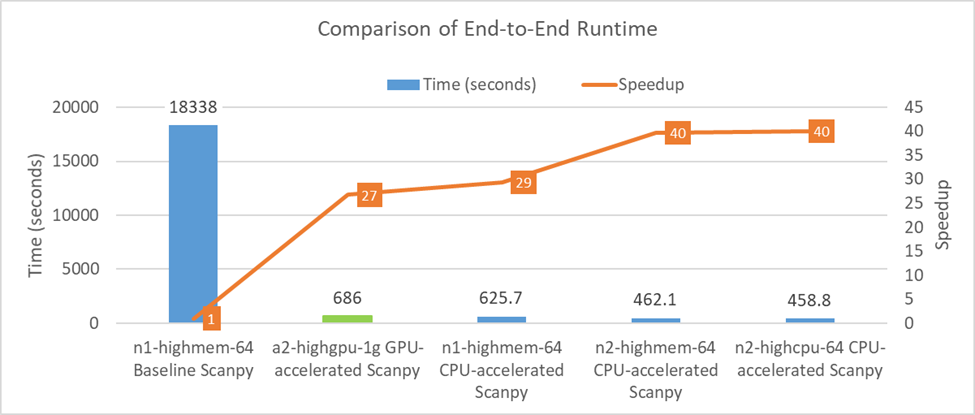
Figure 2. Execution time and speedup for scRNA-seq analysis of 1.3 million mouse brain cells on various GCP instances. The chart uses (1) published execution time of baseline Scanpy on a single CPU instance (n1-highmem-64) and GPU-accelerated Scanpy on a single GPU instance (a2-highgpu-1g), and (2) measured2 execution time of CPU-accelerated Scanpy on single instances of two generations of CPU instance types (n1-highmem-64, n2-highmem-64 and n2-highcpu-64). In addition, the line graph shows the speedup over baseline Scanpy running on n1-highmem-64 instance.
How Was the Data Science Pipeline Accelerated?
Detailed below is a brief summary of the steps we took to improve the performance of this pipeline:
- To increase the efficiency of data preprocessing, we used a warm file cache and multithreading using Numba, a just-in-time (JIT) compiler. This improved the baseline preprocessing performance by more than 70x.
- We also used the Intel® Extension for Scikit-learn* that has efficient implementations of K-means clustering, KNN (K Nearest Neighbor), and PCA (Principal Component Analysis).
- Scanpy originally used scikit-learn’s tSNE (t-distributed Stochastic Neighbor Embedding) implementation that was inefficient for Intel Xeon processors. We achieved nearly 40x speed-up of tSNE by building an efficient implementation through:
- A shared-memory parallel implementation of the Barnes-Hut algorithm
- Parallelization of quadtree building, sorting, and summarization steps using Morton codes
- Continuing our efforts, we optimized (Uniform Manifold Approximation and Projection) by:
- Converting the Python* source code to C++
- Creating an efficient AVX512/AVX2 based implementation for pseudo random number generator
- Using Intel® oneAPI Math Kernel Library (oneMKL) for the eigenvalue computation step
- As part of our collaboration, Katana Graph provided efficient implementations of the Louvain and Leiden community detection algorithms that were integrated into the pipeline.
These developments significantly reduce the time it takes to analyze large datasets, allowing researchers to complete their work 40x faster than baseline on Xeon and 1.5x faster than an NVIDIA* A100 GPU.
Conclusions
Single-cell analysis has applications in many areas: oncology, microbiology, neurology, reproduction, immunology, digestive and urinary systems. Hopefully, reduced working time will allow for a much deeper understanding of different cells, paving the way for medical advances that could have great collective benefits. We are working on further refining the the scRNA-seq analysis pipeline. Specifically, our efforts are focused on making further improvements in the tSNE, UMAP, and Leiden steps.
Configuration Details
GCP n1-highmem-64: 1-instance GCP n1-highmem-64: 64 vCPUs (Skylake), 416 GB total memory, bios: Google, ucode: 0x1, Ubuntu* 20.04, 5.13.0-1024-gcp
GCP n2-highmem-64: 1-instance GCP n2-highmem-64: 64 vCPUs (Ice Lake), 512 GB total memory, bios: Google, ucode: 0x1, Ubuntu 20.04, 5.13.0-1024-gcp
GCP n2-highcpu-64: 1-instance GCP n2-highcpu-64: 64 vCPUs (Ice Lake), 64 GB total memory, bios: Google, ucode: 0x1, Ubuntu 20.04, 5.13.0-1024-gcp